Search results
Search for "molecular dynamics simulation" in Full Text gives 12 result(s) in Beilstein Journal of Organic Chemistry.
Synthesis of ether lipids: natural compounds and analogues
Beilstein J. Org. Chem. 2023, 19, 1299–1369, doi:10.3762/bjoc.19.96

- functions of ether lipids offer an interesting overview of the current knowledge of the effect of plasmalogen and ELs on membrane properties at a molecular scale. Shortly, according to molecular dynamics simulation, PE-plasmalogens form thicker, compressed and rigid bilayers when compared to PE-diacyl






























































































Computational model predicts protein binding sites of a luminescent ligand equipped with guanidiniocarbonyl-pyrrole groups
Beilstein J. Org. Chem. 2022, 18, 1322–1331, doi:10.3762/bjoc.18.137

- distribution; this approach captures ion-size effects and yields energy grids that are directly comparable to molecular dynamics simulation data. Accordingly, the 14-3-3ζ environment was scanned with the GCP and lysine ligands separately in Epitopsy using 150 rotations and a grid resolution of 0.4 Å to







1,2,3-Triazolium macrocycles in supramolecular chemistry
Beilstein J. Org. Chem. 2019, 15, 2142–2155, doi:10.3762/bjoc.15.211

- concentration (10−5 mol/L) in a competitive aqueous–organic CHCl3/CH3OH/H2O mixture (45:45:10, v/v/v). Fluorescence spectroscopy measurements supported by molecular dynamics simulation data have revealed that the smaller macrocyclic rings move from the core-substituted PDI motif to the two 1,2,3-triazolium

















Archangelolide: A sesquiterpene lactone with immunobiological potential from Laserpitium archangelica
Beilstein J. Org. Chem. 2019, 15, 1933–1944, doi:10.3762/bjoc.15.189
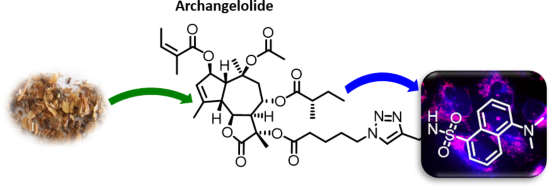
- . Interestingly, we found that neither archangelolide nor its dansyl conjugate did exhibit cytotoxic effects in contrast to the structurally closely related counterparts trilobolide and thapsigargin. We explain this observation by a molecular dynamics simulation, in which, in contrast to trilobolide
- that this SL does not act as SERCA inhibitor. Therefore, we proceeded to confirm this hypothesis by a molecular dynamics simulation study. Molecular dynamics simulation of compounds 1 and 2 with SERCA The binding cavity for thapsigargin and compound 2 in the SERCA protein lies in the transmembrane












Stereo- and regioselective hydroboration of 1-exo-methylene pyranoses: discovery of aryltriazolylmethyl C-galactopyranosides as selective galectin-1 inhibitors
Beilstein J. Org. Chem. 2019, 15, 1046–1060, doi:10.3762/bjoc.15.102





6’-Fluoro[4.3.0]bicyclo nucleic acid: synthesis, biophysical properties and molecular dynamics simulations
Beilstein J. Org. Chem. 2018, 14, 3088–3097, doi:10.3762/bjoc.14.288

- = −1.5 to −3.7 °C). Molecular dynamics simulation on the nucleoside and oligonucleotide level revealed the preference of the C1’-exo/C2’-endo alignment of the furanose ring. Moreover, the simulation of duplexes with complementary RNA disclosed a DNA/RNA-type duplex structure suggesting that this
- finding indicates that the 6’F-bc4,3 modification is more a DNA mimic than an RNA mimic. In accordance with this were the results obtained from the molecular dynamics simulation of the duplexes where the sugar pucker preferably adopted a Southern conformation (C1’-exo, C2’-endo). Furthermore the









Computational methods in drug discovery
Beilstein J. Org. Chem. 2016, 12, 2694–2718, doi:10.3762/bjoc.12.267

- ], GROMACS [192] and AMBER [120]. The typical time-scale of a molecular dynamics simulation is in the order of nanoseconds to microseconds. However to capture biologically important conformational transitions, frequently it is important to probe dynamics in the order of milliseconds. Simulating on that










Physical properties and biological activities of hesperetin and naringenin in complex with methylated β-cyclodextrin
Beilstein J. Org. Chem. 2015, 11, 2763–2773, doi:10.3762/bjoc.11.297

- hesperetin with cyclodextrins (β-CD and DM-β-CD) were theoretically investigated by molecular dynamics simulation. The free energy values obtained suggested a more stable inclusion complex with DM-β-CD. The vdW force is the main guest–host interaction when hesperetin binds with CDs. The phase solubility
- physical properties and biological activities of hesperetin and naringenin through complexation with cyclodextrins. Computational tools (molecular dynamics simulation) were adopted to first predict the stability of flavanones/CDs inclusion complexes. Consequently, the experimental phase solubility and
- values showed the same trend with values from molecular dynamics simulation that complexing with DM-β-CD was more effective than with β-CD, and the values obtained were in good agreement with the previous report [42]. These results suggested that both flavanones bind to and interact with DM-β-CD stronger








Binding mode and free energy prediction of fisetin/β-cyclodextrin inclusion complexes
Beilstein J. Org. Chem. 2014, 10, 2789–2799, doi:10.3762/bjoc.10.296

- complexes. In addition, the quantum mechanics calculations with M06-2X/6-31G(d,p) clearly showed that both solvation effect and BSSE correction cannot be neglected for the energy determination of the chosen system. Keywords: cyclodextrin; fisetin; flavonoid; MM-PBSA; molecular dynamics simulation; QM-PBSA








Structure elucidation of β-cyclodextrin–xylazine complex by a combination of quantitative 1H–1H ROESY and molecular dynamics studies
Beilstein J. Org. Chem. 2013, 9, 1917–1924, doi:10.3762/bjoc.9.226

- clear. Thus, two geometries for the β-CD-xylazine complex can be assumed (Figure 5). We established the structure of the complex using this ROESY data and molecular dynamics simulation studies in vacuum. Earlier, we established the structure of a fexofenadine-α-CD complex [20] on the basis of molecular
- . An iteration step of 1 fs was used and conformations were recorded after every 10 iterations with 4000 steps of equilibration. Molecular dynamics simulation results provided evidence that complexation of β-CD and xylazine is energetically favored. In both the cases, the total potential energy of the
- of two ROESY peaks and r1 and r2 are distances between interacting protons in a given ROESY spectrum. To see whether this relation can be useful for the study of CD complexes, we performed a molecular dynamics simulation of the aspartame-β-CD complexation from the wider side. From the interproton











Peptides presenting the binding site of human CD4 for the HIV-1 envelope glycoprotein gp120
Beilstein J. Org. Chem. 2012, 8, 1858–1866, doi:10.3762/bjoc.8.214

- above. Absorbances (A) were read at 492 nm, and corrected for the respective blanks (wells without X5). All binding assays were performed at least twice, each time in duplicate. MD simulation Computational analysis included modelling of peptide–gp120 complexes followed by molecular dynamics simulation







An easily accessible sulfated saccharide mimetic inhibits in vitro human tumor cell adhesion and angiogenesis of vascular endothelial cells
Beilstein J. Org. Chem. 2012, 8, 787–803, doi:10.3762/bjoc.8.89











